利用数据库数据进行定制化的生信分析
让您的文字更出彩
|
ChIP-Seq测序是将染色质免疫共沉淀(Chromatin Immunoprecipitation,ChIP)与二代测序相结合的表观遗传研究技术,能够高效地在全基因组范围内对DNA和蛋白的相互作用进行检测,通常用于转录因子结合位点或组蛋白特异性修饰位点的研究。
ChIP-seq Assay Kit for Histone Methylation(Laboratorytalk,2016) 我们的优势1. 定制化分析策略:根据不同测序方案和测序目标,定制化选择比对算法、Peak Calling算法以及注释用数据库区域信息; 2. 强大的自研能力:拥有已发表的自主开发Peak Calling算法(Wang et al., BMC Bioinformatics, 2010)和定位注释系统以及一系列下游分析工具; 3. 全面的数据库整合:不断更新基因组数据库并进行多数据库多版本整合,获得准确的Peak定位信息与注释; 4. 强大的组学联合分析能力:将ChIP-Seq与甲基化测序、转录组测序以及全基因组测序等技术进行结合,将单一的蛋白结合数据更进一步拓展。 样本要求注:前期免疫共沉淀(ChIP)实验需客户自主完成,我们提供测序和数据分析服务。 ChIP DNA样品要求: 1. DNA总量≥20ng,浓度≥1ng/μL(Qubit检测),体积要求20-100μL; 2. OD260/280介于1.8-2.0之间; 3. Agilent 2200质检合格,DNA样本主峰范围在100-500bp; 4. 电泳检测无明显RNA污染,基因组条带清晰、完整,无降解; 5. 送样时请标记清楚样品编号,管口使用Parafilm膜密封; 6. 样品保存期间切忌反复冻融; 7. 送样时请使用干冰运输。 实验流程
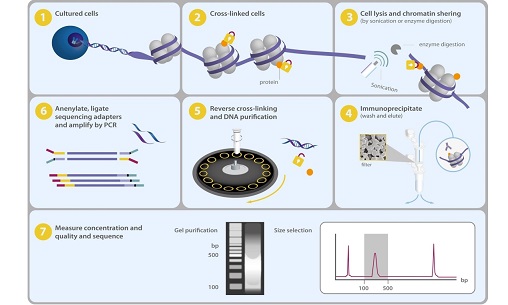
1. 细胞交联固定:甲醛处理细胞,将目标蛋白与染色质连结起来; 2. DNA超声打断:将基因组DNA打断至100-500bp; 3. 免疫共沉淀:添加与目标蛋白特异性结合的抗体,形成免疫沉淀复合物; 4. DNA片段回收:去交联,纯化DNA; 5. 文库构建:PCR扩增构建文库; 6. 上机测序:建议使用Hiseq或NovaSeq测序平台,数据量6Gb。 数据分析流程
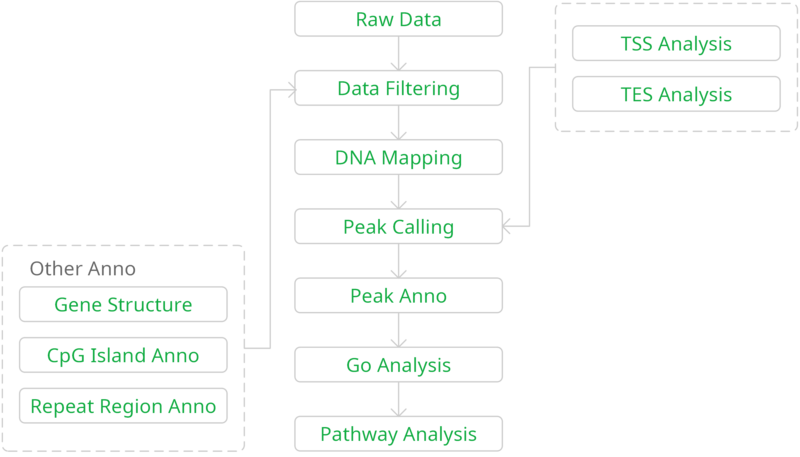
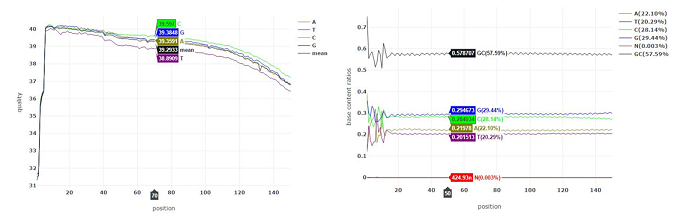
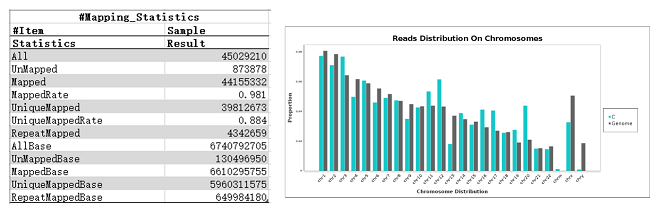
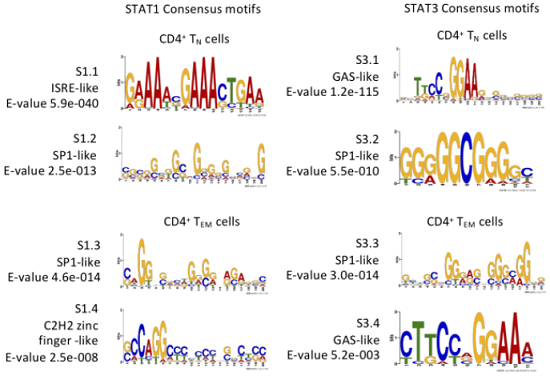
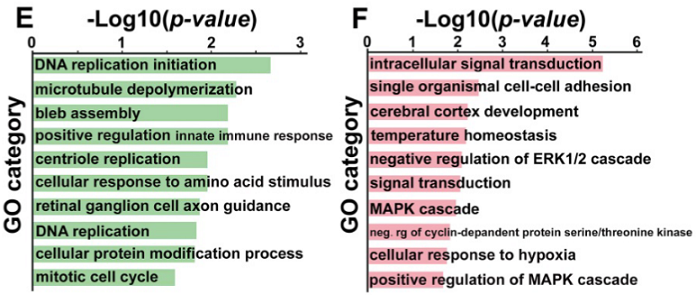
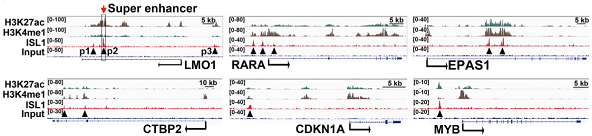
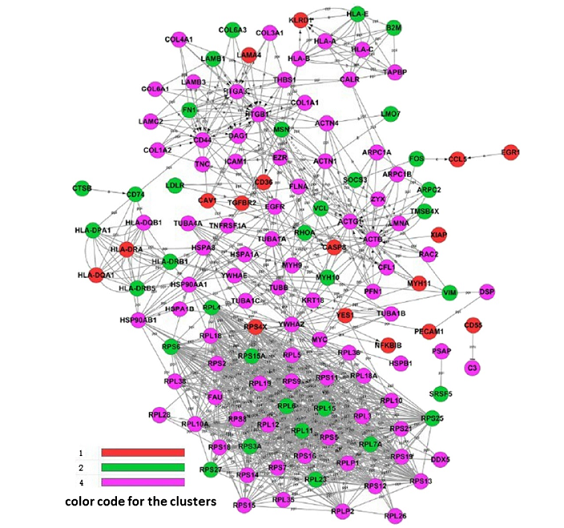
结果示例1、原始数据质量控制采用Fastp软件对下机原始数据中低质量序列,未检测序列,接头序列进行过滤,并对于过滤前后数据的GC比值,碱基质量,长度分布,接头留存,Duplication比率等指数进行分析。图1中部分展示了raw data质控结果。 碱基质量结果图 注:左图横坐标代表碱基位点,纵坐标代表碱基质量值,不同颜色曲线代表不同碱基在每条read上的质量值;右图横坐标代表碱基位点,纵坐标代表碱基含量比值,不同颜色曲线代表不同位点各碱基含量。 2、DNA基因组比对(DNA Mapping)采用Bowtie2算法将过滤后的数据比对到基因组上,得到基因组比对的bam文件,并基于bam文件进行信息统计,得到基因组比对率、reads在染色体上的分布情况。 DNA mapping结果图 注:左图为reads在基因组上的比对情况;右图为reads在染色体上的分布情况,灰色柱子表示每条染色体上碱基数占基因组的比例,绿色柱子表示比对到染色体上reads的碱基数占基因组的比例。 3、Peak Calling和Peak注释(Peak Annotation)采用MACS2算法对DNA Mapping后的bam文件进行peak峰的检测,得到ChIP样本中转录因子结合或组蛋白修饰的富集区域,即peak区域。并采用Chipseeker算法对peak进行注释,得到peak对应的基因以及所在的基因结构,包括启动子、外显子、内含子、基因间区等。此外,我们还可以采用deeptools软件对目标区域内测序序列的覆盖情况进行绘图,得到reads在peak区域或基因结构上的分布情况。 peak数量及其在基因结构上的分布情况 Twohig JP et al., Nat Immunol, 2019 注:左图为不同分组peak在基因组结构上的占比情况,右图为不同分组peak的总数量。 4、Motif分析(Motif Analysis)采用HOMER/MEME算法以及JAPSAR数据库,对peak区域进行motif分析,得到潜在的motif特征序列。 特定转录因子结合motifs的富集情况 Twohig JP et al., Nat Immunol, 2019 注:MEME与STAMP/Jaspar软件共同对STAT1和STAT3预测出来的binding Motif。 5、功能分析(GO Analysis)信号通路分析(Pathway Analysis)分别采用NCBI/UNIPROT/SWISSPROT/AMIGO等GO数据库,以及KEGG数据库对Peak Annotation的相关基因进行功能分析和信号通路分析,得到这些基因群体所显著性富集的GO条目和Pathway条目。 GO分析结果 Zhang QT et al., Theranostics. 2019 注:左图为下调基因所显著富集的GO条目(Top 10),右图为上调基因所显著富集的GO条目(Top 10)。 1、IGV截图采用igvtools算法将基因组比对后bam文件转换为tdf格式,通过IGV软件对目标基因附近的peak分布情况进行可视化展示。 ISL1靶基因处peak分布情况 Zhang QT et al., Theranostics. 2019 注:该图展示了ISL1靶基因附近H3K4me1、H3K27ac和ISL1的peak分布情况,箭头表示结合的peak。 2、基因间相互作用关系网络(Gene-Act-Network Analysis)以显著性GO Analysis和显著性Pathway Analysis的和组蛋白相关的表型基因为研究目标,采用KEGG数据库基因间关系注释,绘制Gene-Act-Network。 基因间相互作用关系网络 Gao et al., Cancer. 2015 注:Profile1、2、4基因集间基因互作网络图。 文献示例[1] Twohig JP, Cardus Figueras A, Andrews R, et al. Activation of naïve CD4+ T cells re-tunes STAT1 signaling to deliver unique cytokine responses in memory CD4+ T cells. Nat Immunol. 2019 Apr;20(4):458-470. (IF=21.809) [2] Mirtschink P, Bischof C, Pham MD, et al. Inhibition of the HIF1α-Induced Cardiospecific HERNA1 eRNA Protects from Heart Disease. Circulation. 2019 Mar 29. (IF=18.88) [3] Han X, Huang H, Gao P, et al. E-protein regulatory network links TCR signaling to effector Treg cell differentiation. PNAS. 2019 Feb 15. pii: 201800494. (IF=9.504) [4] Zhang QT, Zhang QQ, Jiang X, et al. Collaborative ISL1/GATA3 interaction in controlling neuroblastoma oncogenic pathways overlapping with but distinct from MYCN.Theranostics. 2019; 9(4): 986–1000. (IF=8.537) [5] Ranjit M, Hirano M, Aoki K , et al. Aberrant Active cis-Regulatory Elements Associated with Downregulation of RET Finger Protein Overcome Chemoresistance in Glioblastoma. Cell Rep. 2019 Feb 26;26(9):2274-2281.e5. (IF=8.032) [6] Gao Y, et al. Single Cas9 nickase induced generation of NRAMP1 knockin cattle with reduced offtarget effects. Genome Biol. 2017 Feb 1;18(1):13. (IF=9.202) |