利用数据库数据进行定制化的生信分析
让您的文字更出彩
|
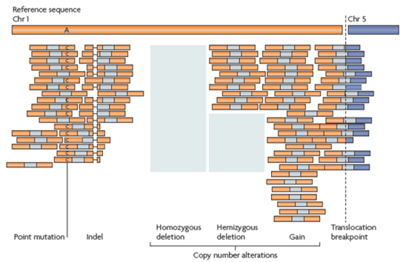
基因组重测序基因组重测序(Re-Sequencing)是对已知基因组序列信息的个体进行测序,可在此基础上对个体或群体进行基因型差异性分析。基因组重测序主要用于辅助研究者发现大量的单核苷酸多态性位点(SNP)、拷贝数变异(Copy Number Variation,CNV)、插入缺失(InDel,Insertion/Deletion)等变异位点,以高效准确获得生物群体的遗传特征,并方便进行全基因组关联性分析(GWAS),在人类疾病和动植物育种研究等方面意义重大。
Types of genome alterations that can be detected by next-generation sequencing Meyerson et al., Nature Reviews Genet,2010 我们的优势1. 重测序分析加速:开创了从任务投递、数据切分到容器多线程的三重调度加速框架,最终实现了重测序分析的大幅度加速,4小时即可完成一个样本的人类重测序分析,较传统的分析方法(68-92小时)提高了十多倍速度; 2. 定制化分析策略:根据不同测序物种和测序方案,定制化选择参考基因组版本、比对算法和注释用数据库区域信息等; 3. 全面的数据库整合:不断更新基因组数据库并进行多数据库多版本整合,获得准确的基因信息与注释; 4. 强大的组学联合分析能力:将基因组重测序与转录组测序和甲基化测序等技术进行结合,将单一的基因变异数据进一步拓展。 样本要求组织样品: 1. 新鲜动物组织干重≥0.5g 2. 新鲜植物组织干重≥2g 3. 新鲜培养细胞数≥4×106个 4. 全血(哺乳动物)≥1ml 5. 全血(非哺乳动物)≥0.5ml DNA样品要求: 1. DNA总量≥2μg,浓度≥20ng/μL,体积要求15-100μL; 2. OD260/280介于1.8-2.0之间; 3. Agilent 2200质检合格,DNA样本主峰范围在100-500bp; 4. 电泳检测无明显RNA污染,基因组条带清晰、完整,无降解; 5. 送样时请标记清楚样品编号,管口使用Parafilm膜密封; 6. 样品保存期间切忌反复冻融; 7. 送样时请使用干冰运输。 实验流程
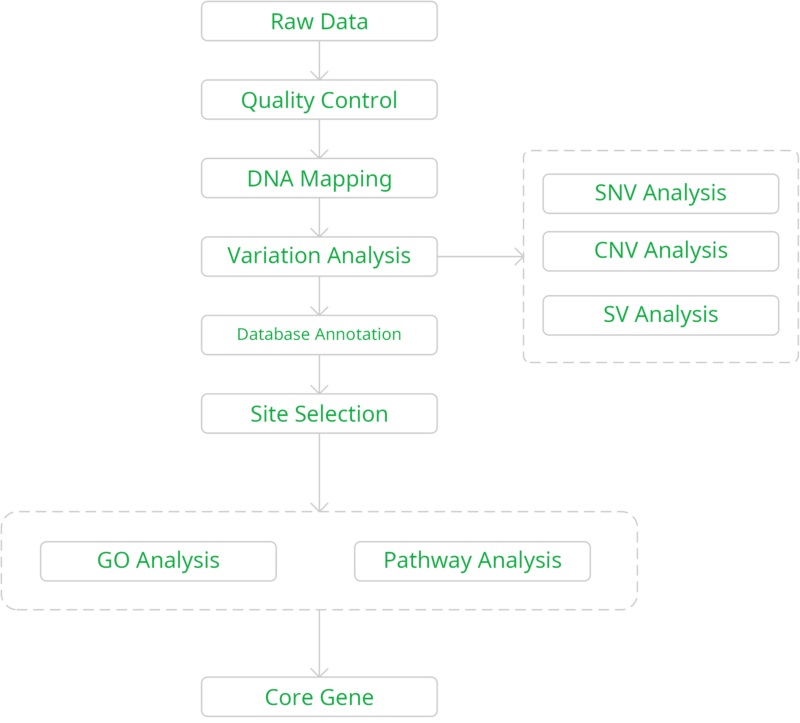
1. 客户样本:新鲜培养细胞数≥4×106个; 2. 提取基因组DNA:DNA总量≥2μg; 3. DNA质控:Agilent 2200质检合格,DNA样本主峰范围在100-500bp; 4. 文库构建:随机引物PCR扩增; 5. 上机测序:测序数据量达到50X覆盖深度,不同物种间存在差异,人一般达到30X。 数据分析流程
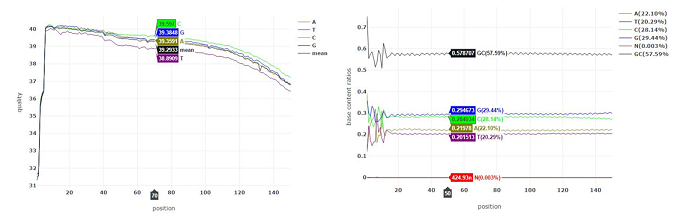
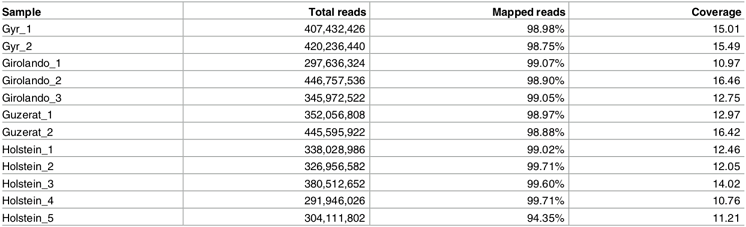
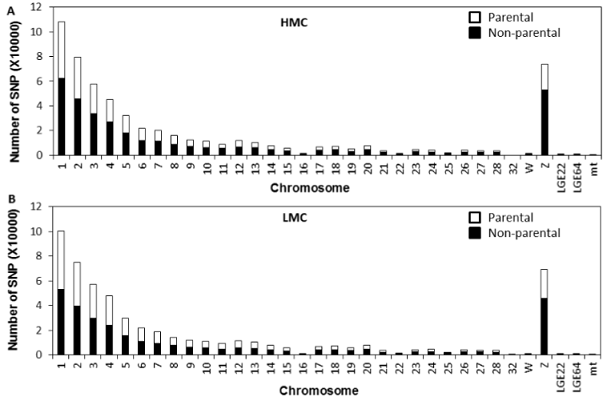
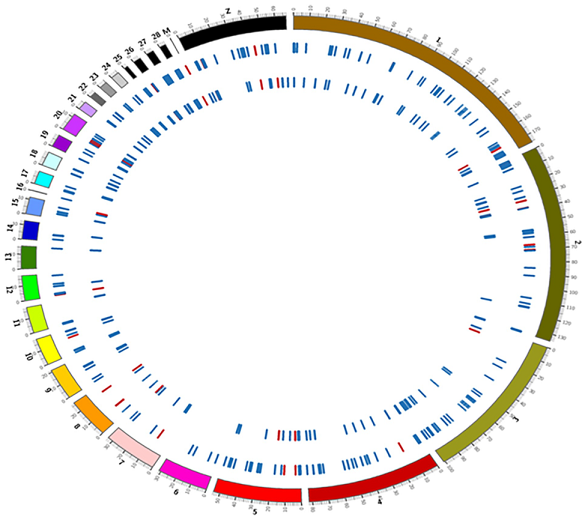
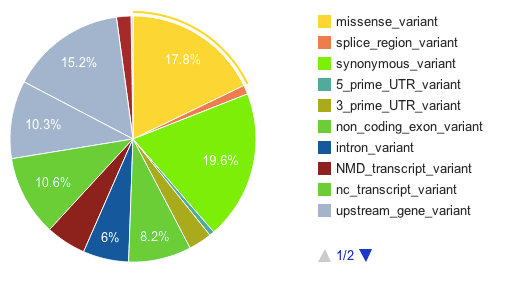
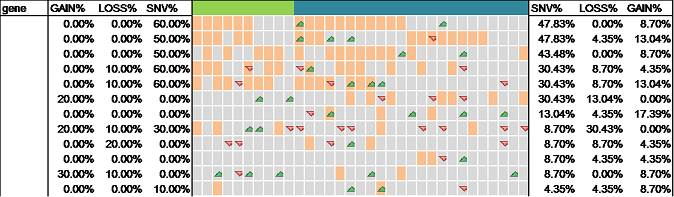
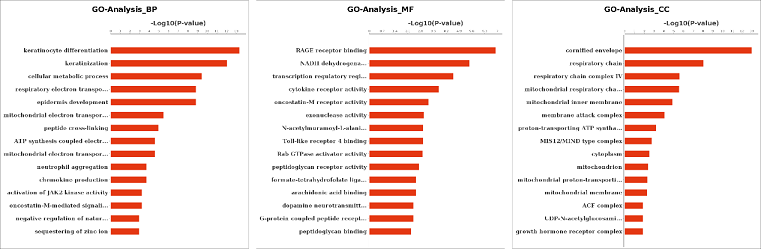
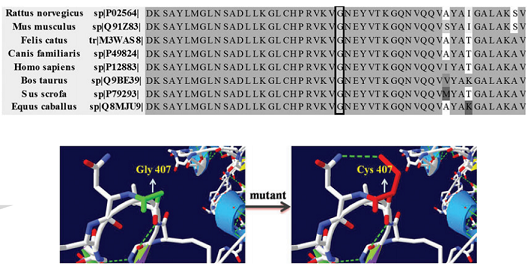
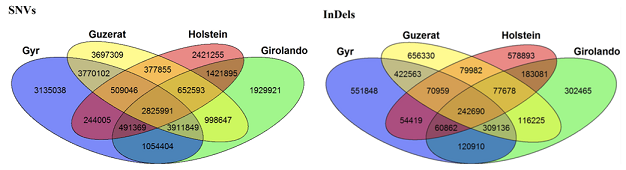
结果示例1、原始数据质量控制采用Fastp软件对下机原始数据进行低质量序列,未检测序列,接头序列的过滤处理,并对于过滤前后数据的GC比值,碱基质量,长度分布,接头留存,Duplication比率等指数进行分析。 碱基质量结果图 注:左图横坐标代表碱基位点,纵坐标代表碱基质量值,不同颜色曲线代表不同碱基在每条read上的质量值;右图横坐标代表碱基位点,纵坐标代表碱基含量比值,不同颜色曲线代表不同位点各碱基含量。 2、DNA基因组比对(DNA Mapping)与质控采用BWA-mem/Bowtie2等算法将质控后的测序数据与基因组进行比对,得到基因组比对的bam文件。并基于bam文件进行信息统计,得到reads在染色体上的分布情况、基因组比对率等信息。 DNA Mapping结果 Yamagishi MEB et al., PLoS One. 2017 注:该图展示了不同品系bull的测序reads的mapping率 3、突变分析采用GATK/Samtools等算法,对基因组比对文件进行突变(包括SNV、InDel)分析,得到突变结果。 call SNP分析结果 Kong HR et al., AJAS. 2018 注:该图展示了不同分组每条染色体SNP的数量 4、拷贝数变异(CNV)分析采用CNVKit算法,对基因组比对文件进行拷贝数变异分析,得到拷贝数变异结果。 CNV区域在全基因组上的分布 Khatri B et al., PLoS One. 2019 注:该图展示了不同品系(HS和LS)的Japanese quail CNV区域在全基因组上的分布情况。外圈为HS品系,内圈为LS品系 5、数据库注释通过VEP对突变分析结果进行注释,获得所有突变位点对应的基因及其变异位点的注释情况。 突变注释结果 注:该图展示了基于数据库鉴定的突变类型,以及不同突变类型数量的占比情况 6、位点筛选以所有的突变注释结果为研究对象,进行突变过滤,得到具有研究意义的突变位点以及其注释信息。 多样本位点筛选 注:该图同时展示了在样本群体中CNV和SNV的变异频率 7、突变基因功能分析(GO Analysis)采用NCBI/UNIPROT/SWISSPROT/AMIGO等GO数据库,对突变基因进行功能分析,得到突变基因所显著性富集的GO条目。 GO分析结果 注:该图从生物过程(BP)、分子功能(MF)和细胞组分(CC)三个方面展示了突变基因显著富集的GO条目(Top 15) 8、突变基因信号通路分析(Pathway Analysis)采用KEGG数据库对突变基因进行信号通路分析,得到突变基因所显著性富集的Pathway条目。 图9 Pathway分析结果 注:该图展示突变基因的Pathway分析结果(Top 15),红色为显著性条目,蓝色为非显著性条目 1、突变位点保守性分析针对missense的突变类型,通过SIFT和Polyphen算法,以位点在物种间的保守性以及位点在蛋白结构中的作用来判断该位点的致病性。 突变位点保守性分析 Guo Q et al., Dna & Cell Biology, 2014 注:多物种MYH7氨基酸序列比对结果表明该突变位点具有高度保守性,并通过软件预测该位点突变前后的蛋白的3D结构 2、韦恩分析(Venn Analysis)将各样本鉴定出的突变位点进行韦恩分析,通过韦恩作图分析的方法,分别得到各样本间独有或共有突变位点,并进一步通过突变注释得到其对应基因,进行GO和Pathway分析。 SNVs和InDels韦恩图 Yamagishi MEB et al., PLos ONE. 2017 注:该图展示了4种品系中鉴定出的SNVs和InDels的Venn分析结果 文献示例[1] Khatri B, Kang S, Shouse S, et al. Copy number variation study in Japanese quail associated with stress related traits using whole genome re-sequencing data[J]. PLoS One. 2019, 14(3):e0214543. [2] Yu Y, Fu J, Xu Y et al. Genome re-sequencing reveals the evolutionary history of peach fruit edibility[J]. Nat Commun. 2018, 9(1):5404. [3] Reimer C, Rubin CJ, Sharifi AR, et al. Analysis of porcine body size variation using re-sequencing data of miniature and large pigs[J]. BMC Genomics. 2018, 19(1):687. [4] Kong H R, Anthony N B, Rowland K C, et al. Genome re-sequencing to identify SNP markers for muscle color traits in broiler chickens[J]. Asian-Australasian Journal of Animal Sciences, 2018, 31(1):13-18. [5] Yamagishi MEB, Chud TCS, CaetanoAR, et al.Single nucleotide variants and InDels identified from whole-genomere-sequencing of Guzerat, Gyr, Girolando and Holstein cattle breeds. PLoS One. 2017, 12(3):e0173954. [6] Linnéa Smeds, Mugal C F, Anna Qvarnström, et al. High-Resolution Mapping of Crossover and Non-crossover Recombination Events by Whole-Genome Re-sequencing of an Avian Pedigree[J]. PloS Genetics, 2016, 12(5):e1006044. [7] He Y, Wang C, Higgins J, et al. MEIOTIC F-BOX Is Essential for Male Meiotic DNA Double Strand Break Repair in Rice[J]. The Plant Cell, 2016, 28(8):1879-93. [8] Wei F, Jie Z, Zhijing L, et al. Development of a RAD-Seq Based DNA Polymorphism Identification Software, AgroMarker Finder, and Its Application in Rice Marker-Assisted Breeding[J]. PLoS One, 2016, 11(1):e0147187. [9] Torkamaneh D, Laroche, Jérôme, Belzile, François, et al. Genome-Wide SNP Calling from Genotyping by Sequencing (GBS) Data: A Comparison of Seven Pipelines and Two Sequencing Technologies[J]. PLoS One, 2016, 11(8). [10] Guo Q, Xu Y, Wang X, et al. Exome Sequencing Identifies a Novel MYH7 p.G407C Mutation Responsible for Familial Hypertrophic Cardiomyopathy[J]. Dna & Cell Biology, 2014, 33(10):699-704. 下一篇Small RNA测序 |